CoVaCS on Galaxy
CoVaCS, Consensus Variant Calling System, is a fully automated system for genotyping and variant annotation of resequencing data produced by second generation NGS technologies. The CoVaCS pipeline integrates cutting-edge tools for variant calling and annotation for whole genome sequencing (WGS), whole-exome sequencing (WES) and target-gene sequencing (TGS) data.
The implementation of CoVaCS on Galaxy performs the following pipeline steps:
Quality control
Quality Trimming
Mapping
Variant calling
Variant selection
Variant annotation
Each step of the pipeline is performed by one or more bioinformatics tools:
Quality control
The quality control of raw reads is performed by FastQC.
- Description:
This step provides quality control report on raw sequence data spotting problems which originate either in the sequencer or in the starting library material. The report gives a quick view of the quality of raw data, making the user aware of any quality problems before making any further analysis
- Galaxy wrapper:
Quality Trimming
The quality trimming step is performed by Trimmomatic
- Description:
Taking into account the data problems found in the previous step, Trimmomatic provides the possibility to optimize the raw reads length. It includes several options to read trimming and filtering.
- Galaxy wrapper:
Mapping
The mapping step is performed by the Burrows-Wheeler Aligner (BWA) software package for mapping sequences against a large reference genome.
- Description:
It uses a Burrow’s Wheeler Transform method to map the reads on the reference genome creating a Sequence/Alignment Map (SAM) file for each sample.
- Galaxy wrapper:
Variant calling
The variant calling step is performed by three different tools: Varsca2, GATK and Freebayes. Each tool gives two different output one for the discovery of Indels and one for SNPs.
Varscan2
- Description:
Varscan2 adopts a series of stringent quality metrics in order to identify putative false positive predictions.
- Galaxy wrapper:
GATK
- Description:
GATK performs local reassembly of the reads to mitigate sequence errors and reconstruct haplotypes using VariantRecalibrator and ApplyRecalibrator for standard CoVaCS implementation and Select filtration wrapper in case of not enough snp or indels error in covacs_VariantRecalibrator.
- Galaxy wrapper:
wrapper VariantRecalibrator | wrapper ApplyRecalibrator | wrapper SelectFiltration
Freebayes
- Description:
Freebayes (Garrison and Marth 2012) is based on a probabilistic haplotype reconstruction algorithm.
- Galaxy wrapper:
Variant selection
- Description:
All the SNPs discovered using these two approaches, are grouped into two separate outputs using two perl script
intersect_snpandintersect_indels: common SNPs (SNPs detected by two or three tools) and unique SNPs (SNPs discovered by only one tool). The same process is applied to Indels generating common Indels and unique Indels files.- Galaxy wrapper:
wrapper covacs intersect SNP | wrapper covacs intersect indels
Variant annotation
- Description:
The variant annotation step, both of common and unique variants, is performed by Annovar (Wang, Li, and Hakonarson 2010). Annovar annotates genetic variants returning:
Gene-based annotation: identify whether SNPs or CNVs cause protein-coding changes and the amino acids that are affected.
Region-based annotation: identify variants in specific genomic regions.
Filter-based annotation: identify variants that are documented in specific databases.
- Galaxy wrapper:
The output comprising the annotated variants can be uploaded and visualized for example on the UCSC genome browser.
CoVaCS reference data
CoVaCS reference data are automatically mounted on Galaxy, selecting ELIXIR-IT Galaxy CoVaCS reference data CVMFS repository during the Instance configuration.
The available reference data are:
Reference Genome indexed for BWA and GATK downloaded from GATK bundle ucsc.hg19.fasta
Annovar Databases
Gene-based annotation:
refGene
Filter-based annotation:
Exac03
1000g2015aug
avsnp150
clinvar_20180603
cosmic70
dbnsfp33a
esp6500_all
kaviar_2015092361
knownGene
mitimpact2
gnomad_genome
Downloaded from Annovar repository using the command
$ perl annotate_variation.pl -downdb -buildver hg19 -webfrom annovar <database_name> humandb
CoVaCS workflows
The first workflow was implemented to run the standard pipeline of CoVaCS (fig.1) starting after the quality control, trimming and mapping.

Fig.1 - CoVaCS standard workflow
The second workflow (fig.2) differs from the previous one since the GATK VariantRecalibrator and ApplyRecalibrator are replaced by the Select filtration wrapper. This workflow has been developed in order to be used by users if enough snp or indels error in VariantRecalibrator.
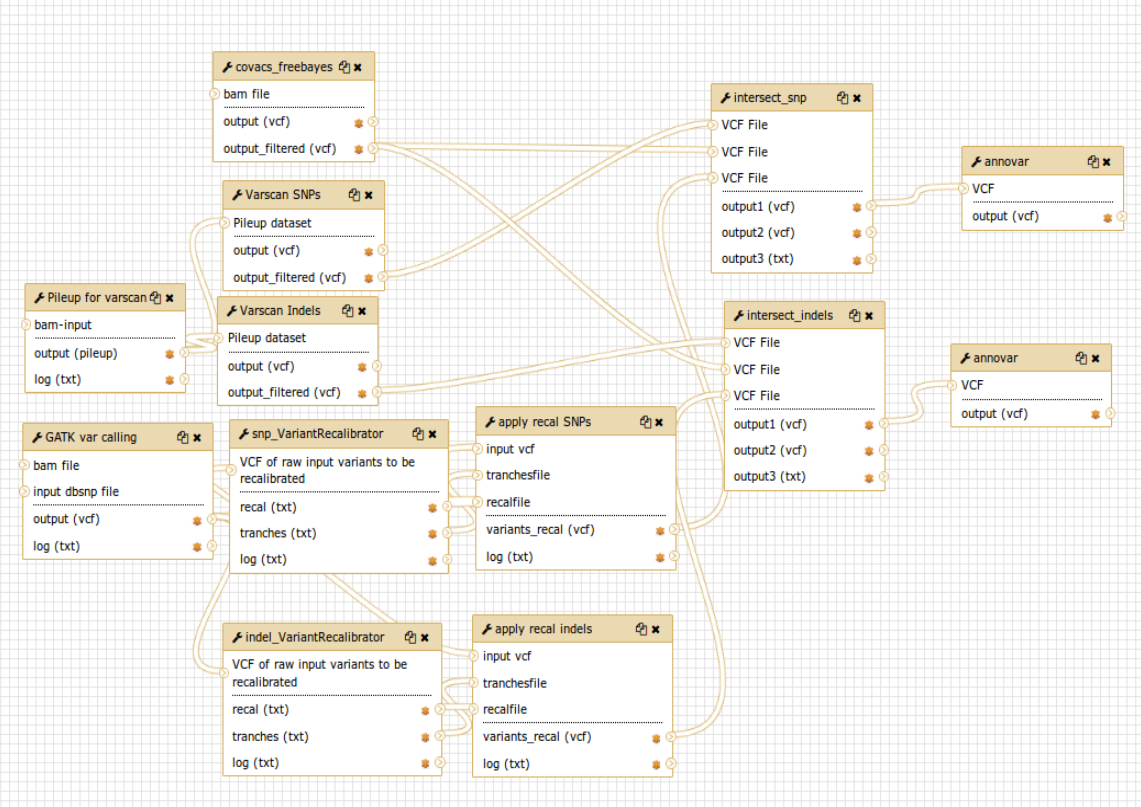
Fig.2 - CoVaCS Select Filtration workflow